Preliminary Results
Important
The preprint is now out! The below results are slightly out of date.
Algorithm outline
At a high level, myloasm uses a string graph approach. Briefly, myloasm:
- aligns reads-to-reads, splits chimeric reads, and estimates coverage per read
- uses polymorphic, strain-specific k-mers (we call them SNPmers) to find true sequence divergence between reads
- obtains a high-resolution overlap graph and finds walks (contigs) with consistent coverage using an annealing-inspired optimization approach
- aligns reads-to-contigs and polishes using partial order alignment (SPOA)
Results
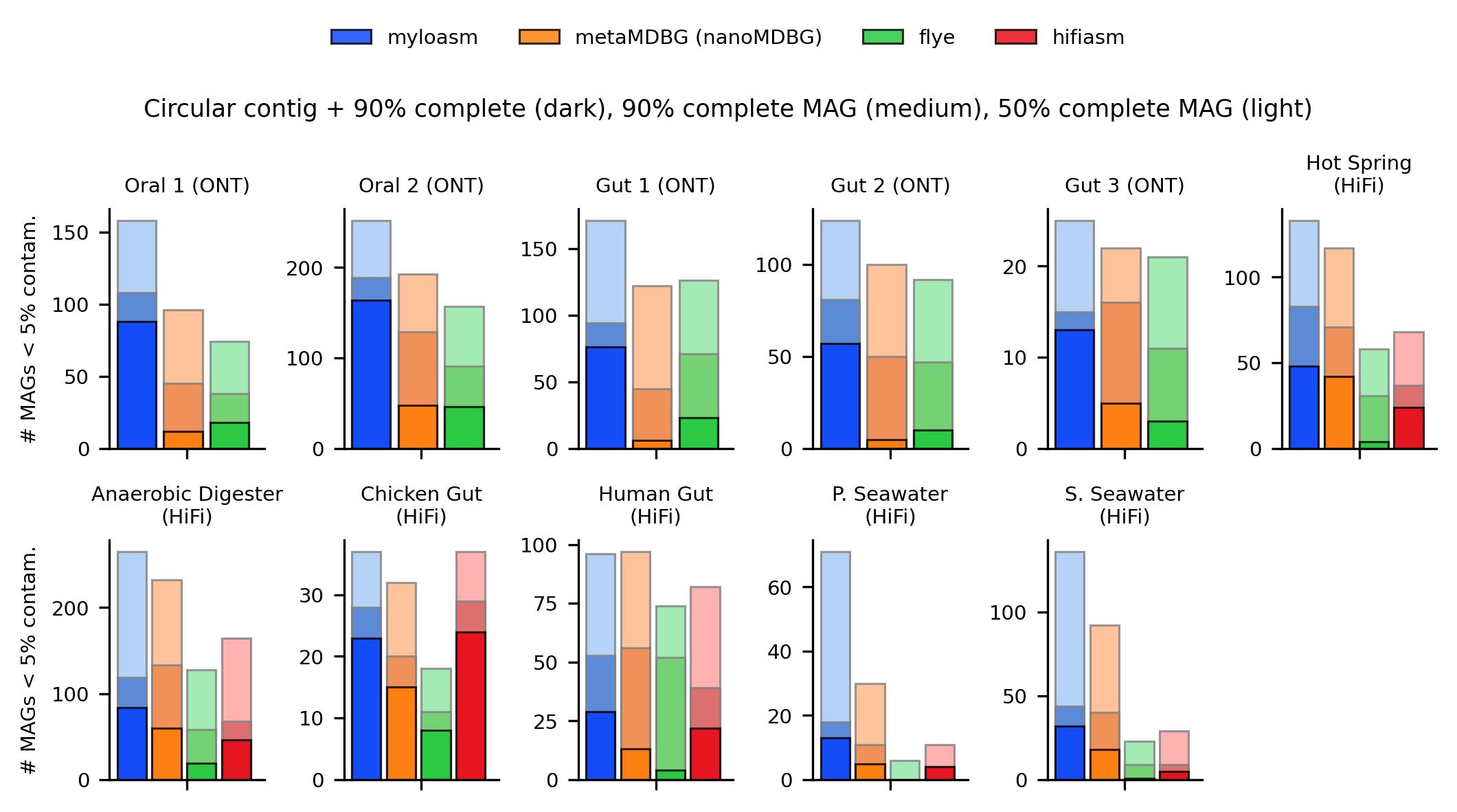
Fig. 1. Metagenome-assembled genome (MAG) recovery for Nanopore R10.4 simplex + PacBio HiFi sequencing datasets. Alignment + binning was done with minimap2 and SemiBin2; results are evaulated with CheckM2.
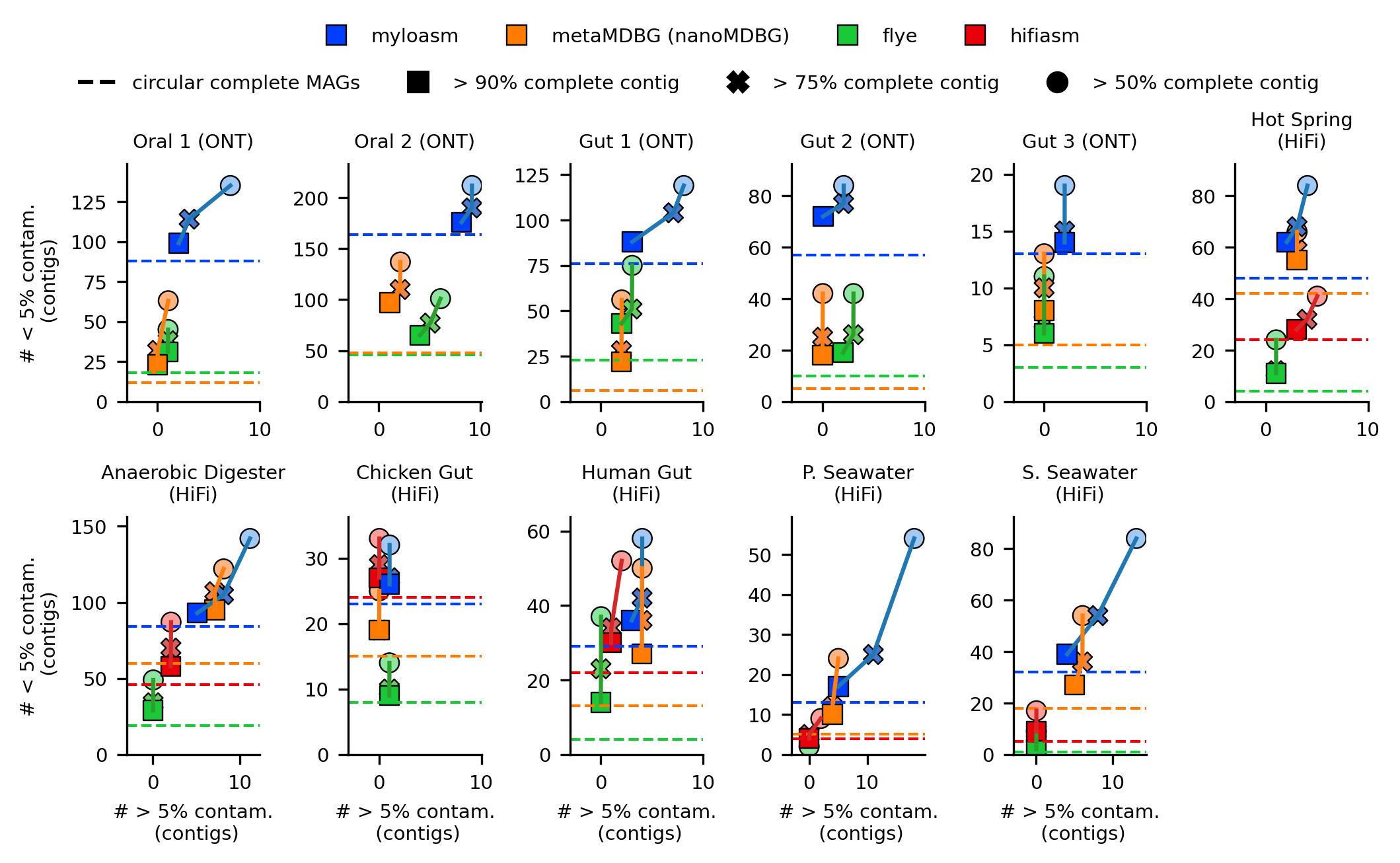
Fig. 2. Completeness + contamination results from CheckM2 on individual contigs for the different assemblers. Dashed line indicates circular complete MAGs (> 90% complete, < 5% contaminated and circular).
Dataset accessions
| Dataset (ONT R10.4) | Accession | Dataset (HiFi) | Accession |
|---|---|---|---|
| Oral 1 (Kiguchi et al.) | DRR582205 | Hot Spring (Kato et al.) | DRR290133 |
| Oral 2 (Kiguchi et al.) | DRR582179 | Anaerobic Digester (Benoit et al.) | ERR10905741 |
| Gut 1 (Minich et al.) | SRR29980972 | Chicken Gut (Zhang et al.) | SRR19683891 |
| Gut 2 (Minich et al.) | SRR29980959 | Human Gut (Gehrig et al.) | SRR15489018 |
| Gut 3 (Minich et al.) | SRR29980980 | P. Seawater (Priest et al.) | ERR4920901 |
| S. Seawater (Sidhu et al.) | ERR9769281 |